Atualização em: 03/08/2022
A Atrofia Muscular Espinhal, também é conhecida como amiotrofia espinhal progressiva. O adjetivo “espinal” também pode ser empregado. Trata-se de uma doença de origem genética, ou seja, não se adquire ela em algum lugar, mas se nasce com ela, alocada no DNA, sendo passada de geração em geração.
Mas como a Atrofia Muscular Espinhal acontece?
Pra responder essa pergunta, a gente precisa saber a causa.
Todos nós possuímos o gene SMN1 – uma cópia herdada do pai e outra da mãe. São justamente mutações nessas cópias que podem causar a AME. Aproximadamente 95% dos indivíduos afetados apresentam a deleção, isto é, perda de uma ou mais unidades que compõem um gene, em ambas as cópias SMN1.
Indivíduos que possuem uma só cópia mutada não terão sintomas da doença. Porém, são portadores mesmo assim. Isso significa que caso eles tenham filhos com outras pessoas que carreguem uma cópia mutada, as chances dessa criança nascer com atrofia muscular espinhal são de 25%.
O SMN1 é responsável por produzir uma proteína fundamental chamada proteína de sobrevivência do neurônio motor. Embora ela esteja presente em todas as células, a proteína é mais importante para o sistema nervoso, sendo a gravidade da doença atrelada à quantidade que o doente consegue produzir. Essa falta acarreta degeneração e perda motora na medula espinhal e do tronco cerebral.
Com os movimentos progressivamente limitados com o avanço da doença, os músculos atrofiam. E é todo tipo de músculo: além dos mais óbvios como braços e pernas, os responsáveis pela deglutição de alimentos e respiração são comprometidos também. Esse triste cenário leva o paciente a precisar de aparelhos pra comer e respirar, infelizmente.
A atrofia muscular espinhal é a segunda doença recessiva letal mais comum em pessoas de pele branca, com recorrência média de 1 para cada 10 mil nascimentos. Dependendo da idade em que ela aparece e sua evolução, a ela é subdividida em tipos:
Tipo 1 – AME infantil ou de Werdnig-Hoffmann
O neurologista alemão Johann Hoffmann cunhou o nome amiotrofia espinhal progressiva em 1893. Dois anos antes disso, o neurologista austríaco Guido Werdnig descreveu o tipo infantil, que é o mais grave de todos, apresentando sintomas ainda na vida intrauterina. Essas crianças normalmente não conseguem passar dos 3 anos de idade. A maior causa de mortandade delas é o subdesenvolvimento do sistema respiratório.
Tipo 2 – AME intermediária
Após o nascimento, os sintomas começam a aparecer e são um pouco menos intensos que na versão anterior. Com cerca de um ano e meio de vida, a criança pode desenvolver a habilidade de sentar, desde que colocadas nessa posição. Andar, entretanto, não conseguem.
Tipo 3 – AME juvenil – Kugelberg-Welander
Os neurologistas suecos Lisa Welander e Erik Kugelberg diferenciaram esta doença das outras distrofias musculares em 1956. Aqui os sintomas aparecem entre o 2º e o 17º ano de vida e os membros superiores são os mais afetados. Precisam de ajudar pra se locomover e fazer coisas do dia-a-dia, mas a progressão da doença é mais lenta.
Tipo 4 – AME adulta
A forma menos grave da doença acomete pessoas entre 30 e 40 anos de idade. Possui a progressão mais lenta de todas as AMEs.
Os medicamentos para AME estão entre os mais caros do mundo e precisam ser administrados o mais rápido possível, juntamente com terapias multidisciplinares.
A impotância do diagnóstico precoce
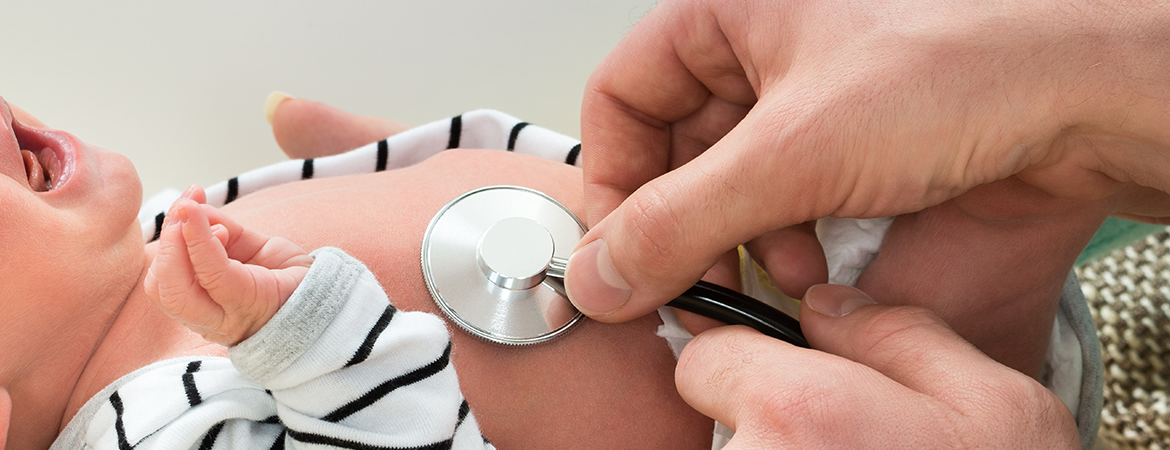
A Atrofia Muscular Espinhal (AME) deve ser diagnosticada o mais rápido possível, pois a intervenção precoce – farmacológica ou não – pode mudar o futuro do paciente e da família, melhorando a qualidade de vida de todos.
Esse é um grande desafio quando se considera um país desigual e de dimensões continentais. A articulação para chegarmos ao diagnóstico precoce exige empenho importante de diferentes atores dentro do nosso sistema de saúde.
O diagnóstico da AME pode ser feito em pacientes sintomáticos ou pré-sintomáticos
Pacientes sintomáticos: Aqueles que já desenvolveram algum sintoma da AME. O diagnóstico parte, geralmente, da suspeita clínica.
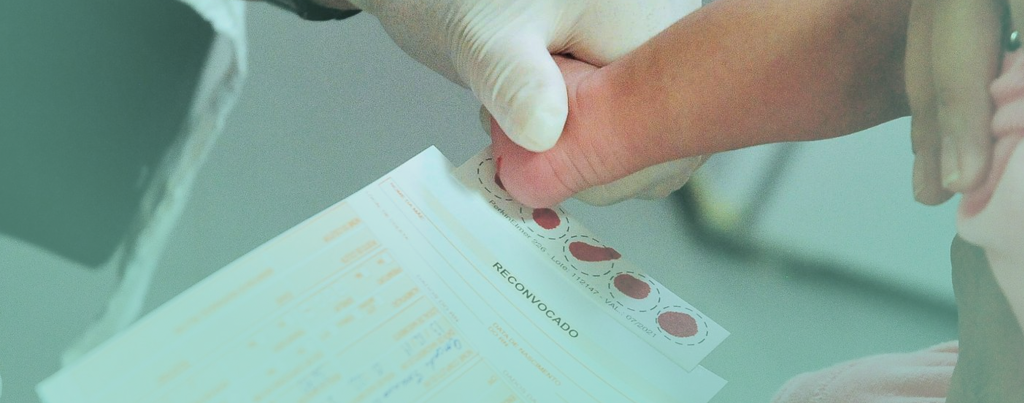
Pacientes pré-sintomáticos: Aqueles que ainda não apresentam sinais ou sintomas de AME. O diagnóstico parte de uma suspeita familiar, em casos de parentes com AME, ou da triagem neonatal por meio do Teste do Pezinho Ampliado, na população em geral.
A triagem neonatal para AME
Aprendizados com projetos piloto de triagem neonatal para AME ao redor do mundo mostram que o diagnóstico precoce é viável considerando aspectos sociais, clínicos e financeiros.
O número de doenças que integram a triagem neonatal, assim como a taxa de cobertura e os métodos laboratoriais variam de acordo com as condições geográficas, econômicas, políticas, demográficas e epidemiológicas de cada país.1-2 No Brasil, o Programa Nacional de Triagem Neonatal (PNTN) contempla seis doenças, mas com a aprovação da Lei 14.154, de maio de 2021, a cobertura será ampliada, e a atrofia muscular espinhal (AME) entrará na quinta etapa do processo de implementação da lei, no Teste do Pezinho Ampliado, o que ainda pode levar anos para ser efetivado.
A aprovação em si já é um grande avanço para a comunidade da AME no Brasil. Porém, acelerar o processo de ampliação das doenças cobertas no PNTN é essencial e pode gerar impacto positivo na vida de muitas famílias, que encontram no diagnóstico precoce uma esperança de que seus bebês possam ser tratados no momento ideal, como indicam diversos estudos realizados ao redor do mundo.
Mãe Que Ama deseja que cada vez mais pessoas tenham acesso ao diagnóstico precoce, a tratamentos imediatos, adequados e acessíveis.
Saiba mais sobre a Atrofia Muscular Espinhal em: Juntos pela AME com Biogen.